massXpert2 User Manual
3 XpertDef: Definition of Polymer Chemistries
After having completed this chapter the reader will be able to accomplish the very first steps needed to use massXpert's features at best: the normal workflow, indeed, is to first make a polymer chemistry definition, in order to be able to edit polymer sequences of that specific definition. The XpertDef module is made available in massXpert by pulling down the menu item from the program's menu. It is possible to start a new polymer chemistry definition from scratch, but it is certainly usually easier to first duplicate a polymer chemistry definition shipped with massXpert and then open that copy and edit it. Please, refer to chapter data-customization, for an explanation of how this is safely done.
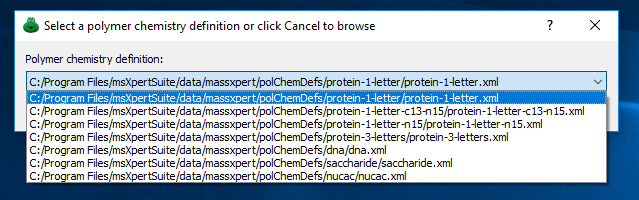
It is possible to immediately select a polymer chemistry definition already registered with the system, or open an arbitrary file by browsing the filesystem (click the button, hidden in this figure, if so desired).
Figure 3.1: select one polymer chemistry definition file. #
To open a polymer chemistry definition, the user may either select one that is already registered with the system, and that appears listed in the drop-down list widget shown in figure Figure 3.1, “select one polymer chemistry definition file.” or click the button so as to open one definition file by browsing the filesystem. In the polymer chemistry definition window that shows up, the user accomplishes two different tasks:
Define the name of the polymer chemistry definition;
Define “singular” data like the left cap and the right cap of the polymer, the ionization rule governing the default ionization of the polymer sequence;
Define the isotopes needed to operate the different polymer chemistry entities (these are “plural” data) ;
Define all the polymer chemistry entities needed to work on polymer sequences (all these are also “plural data”) .
The definition of the isotopes and of all the chemical entities belonging to a given polymer chemistry are collectively called a polymer chemistry definition. The polymer chemistry definition window that shows up is shown in figure Figure 3.2, “ Polymer chemistry definition window.”.
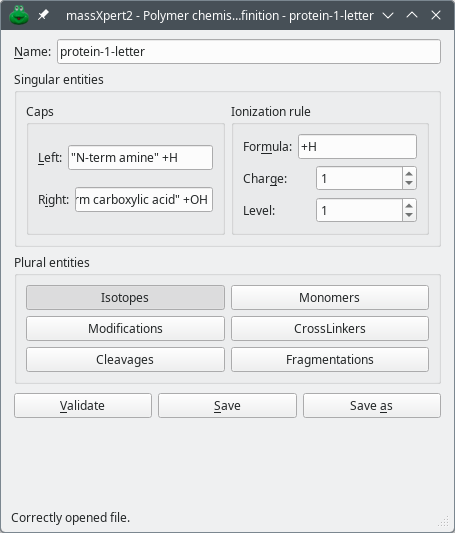
All the polymer chemistry entities are defined in this window. the different buttons dealing with isotopes, monomers, modifications, cross-linkers, cleavage and fragmentation specifications open up specific dialogs (see below).
Figure 3.2: Polymer chemistry definition window. #
3.1 The isotopes #
The definition of the isotopes is performed through the user interface shown in figure Figure 3.3, “ Isotopes definitions” ( button in the polymer chemistry definition window). In this dialog, the user defines all the isotopes that collectively form the chemical elements (at least one isotope per atom, logically). In the table view, each row corresponds to an isotope.
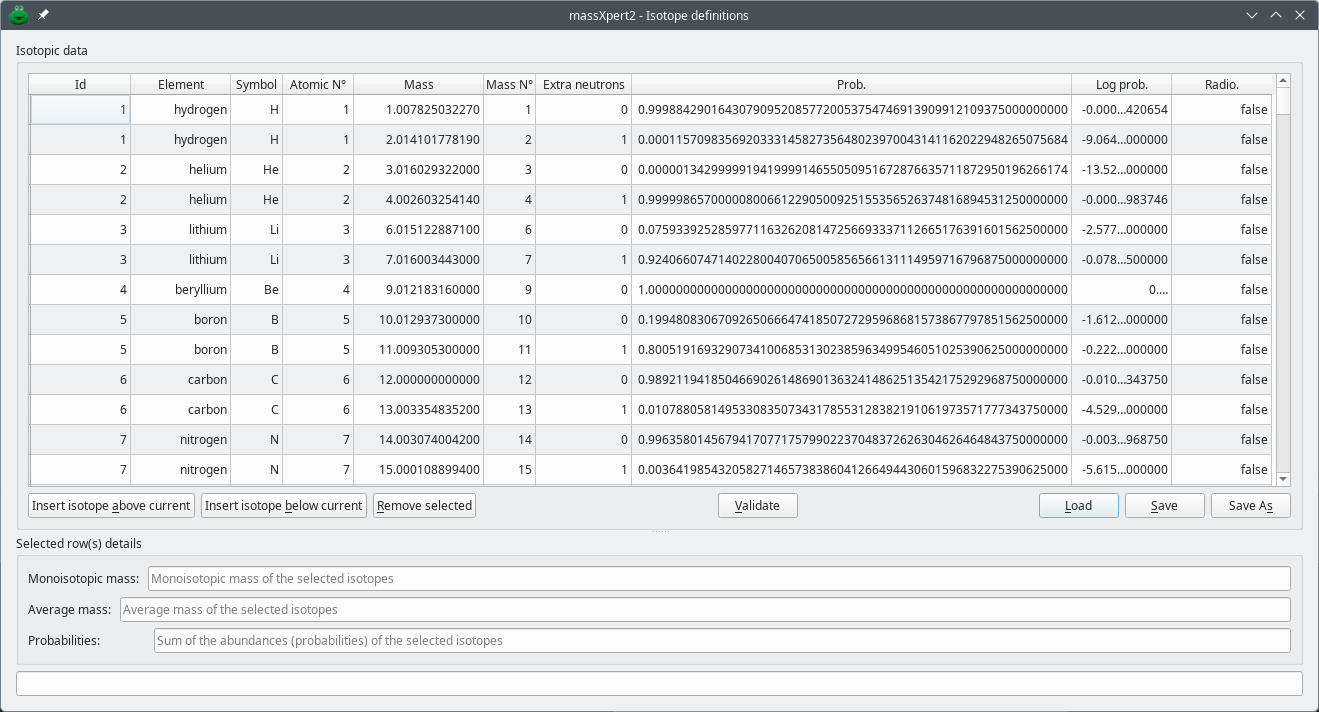
Each row of the table view corresponds to an isotope. For any given element, there must be at least one isotope.
Figure 3.3: Isotopes definitions #
The table view is editable. Each cell, when double-clicked, switches to an edit mode in which the user might enter a value. A series of buttons below the table view trigger various actions. To add a new isotope, either above or below the currently selected one, click of the two Insert isotope... buttons. To remove isotopes, select them and click the Remove selected button. The three buttons on the right hand side of the window, allow loading, saving and saving with a new file name the isotopes currently displayed in the table view.
When the user selects one or more rows of the table, the program updates some data specific to the selected isotopes. As long as the selected isotopes all have the same chemical element, they are taken into account for the calculation of the average mass (Average mass). The Probabilities line edit widget displays the sum of the abundances of the selected isotopes (only if these are of the same chemical element).
When the user finishes editing the probability of a given isotope, the program automatically updates that probability's logarithm value and refreshes the corresponding Log prob. cell on the same row.
3.2 The polymer chemical entities #
Once the atoms have been properly defined (note that such atoms are already available in the distributed package), it is possible to start entering data for the other polymer chemical entities (figure Figure 3.2, “ Polymer chemistry definition window.”. These are often defined using chemical formulas, which explain why it is necessary to first define the atoms.
The following are the data that need to be entered so as to obtain a usable polymer chemistry definition:
The polymer chemistry definition's name: protein-1-letter;
The chemical capping reactions that should happen on the left end and on the right end of the polymer sequence:
+H: left capping of the polymer sequence. proteins are capped at the n-terminal end with a proton;
+OH: right capping of the polymer sequence. proteins are capped at their c-terminal end with a hydroxyl group;
The ionization rule describes the manner in which the polymer sequence should be ionized by default, when the mass is calculated. This rule actually holds three elements:
+H: chemical reaction that ionizes the polymer sequence. in the example, all the polymer sequences of polymer chemistry definition “protein-1-letter” are protonated by default;
1: charge that is brought by the chemical agent ionizing the polymer (the formula above). In the example, a protonation reaction brings a single positive charge.
1: ionization level, that is, the number of times that the ionization (above) must be performed by default on any polymer sequence of this chemistry definition. In this example, monoprotonation is set as the default ionization rule.
At this point, time has come to deal with “plural” data. the first chemical entities to deal with are monomers.
3.2.1 The monomers #
The monomers are the constitutive blocks of the polymer sequence. in the massxpert2's jargon, “monomer” stands not for the molecule that may be used to perform a polymer synthesis; it stands for this molecule less the chemical group(s) that were eliminated upon polymerization. In the case of the biological polymers, the creation of chemical link between two monomers invariably leads to the loss of a water molecule (that is also called a condensation reactions in organic chemistry).
Click onto the button, which triggers the opening of the dialog window shown in figure Figure 3.4, “ Monomer definition”.
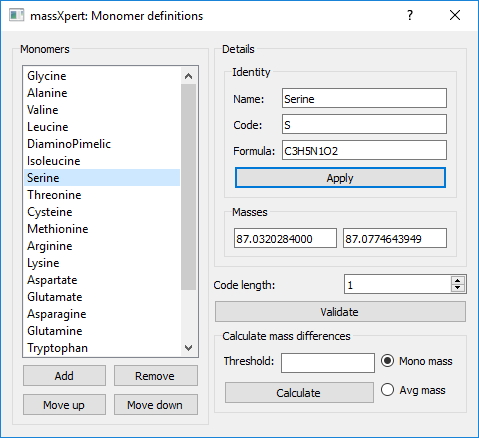
Each monomer is defined with a name, a code and a chemical formula.
Figure 3.4: Monomer definition #
The way this dialog is operated is similar to what was described for the atom definition, unless it is simpler, because monomers are non-deep objects: there are no contained objects. One data element is critical: the number of characters that might be used to define the code of the element cannot be greater than the value entered in the code length spinbox widget[4].
The fundamental rule is the following:
Warning
The first character of a monomer code must be uppercase, while the remaining characters (if any) must be lowercase. That means that—if code length is 3—“a”, “al”, “ala” would be perfectly fine, while “alan”, “al”, “a”, “ala” would be wrong.
Each time a formula is either displayed by selecting a new monomer in the list or modified by editing it in its line edit widget, the monoisotopic and average masses are recalculated.
As of version 2.3.5, it is possible to calculate the mass difference between any two monomers in the definition. This is useful, for example, to grasp the resolution and mass accuracy requirements for a given polymer definition. The user sets a threshold to filter the results (in the example, that mono mass threshold was set to 1. The results of such a calculation are displayed in figure Figure 3.5, “ Monomer mass differences”.
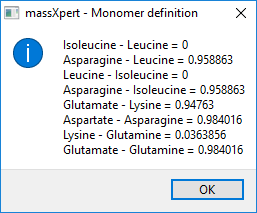
The mass difference between any two monomers in the definition is computed and displayed only if it is less or equal to a threshold (see figure Figure 3.4, “ Monomer definition”
Figure 3.5: Monomer mass differences #
After addition of the monomers it is always a good idea to validate them by clicking .
3.2.2 The modifications #
Polymers are often either chemically or biochemically modified. in nature, biopolymers are modified more often than not. Some of the more common modifications in the protein reign are phosphorylation, acetylation and methylation, for example. Nucleic acids are modified with a sheer number of chemical modifications, saccharides also. The massxpert2 software provides entire freedom to define any number of intelligent modifications, that is, modifications with any chemical formula but also that are knowledgeable of what monomers they can modify. indeed, it would make no sense to phosphorylate a glycyl residue in a protein, for example.
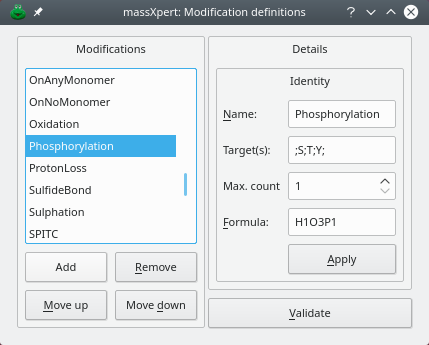
Each modification is defined with a name, targets, a count number and a chemical formula.
Figure 3.6: Modification definition #
Click modifications to open the dialog window shown in figure Figure 3.6, “ Modification definition”. In the example shown, the phosphorylation modification is being defined. a modification is defined by:
The name of the modification: name;
A “;”-separated list of codes of the monomers that might be modified by this modification: targets;
The maximum number a given monomer might be modified with this modification (max. count). This feature is essential when working on methylation of proteins, for example, with arginyl and lysyl residues being multi-methylated;
The formula that defines the modification chemical reaction, as explained in Section 1.1, “On Chemical Formulæ and Chemical Reactions” (formula). Note that, in the example of the figure, for phosphorylation, the formula is a net formula. That formula could be more explicit by entering -h+h2po3. The net formula is thus the one visible on the figure.
The phosphorylation reaction can thus be read like this: —“the polymer looses a proton and gains h2po3”. the phosphorylation is being defined as having s;t;y targets only, that means that when the user will try to modify non-seryl or non-threonyl or non-tyrosinyl monomers, the program will complain that these monomers are not targets of phosphorylation. there is, however, and for maximum flexibility, the possibility to override these target-limiting data when modifying monomers. When a monomer is modified with this modification, its masses will change according to the net mass of this phosphorylation “reaction”.
3.2.3 The cross-linkers #
Polymers are often either chemically or biochemically modified by interconnecting monomers from the same polymer sequence. In the protein reign, one classical example of intra-sequence cross-linking is the formation of disulfide bonds. Another wonderful example is the formation of the fluorophore in the fluorescent proteins: there is a chemical reaction involving the side chains of three consecutive residues going on, resulting in the formation of a complex intra-sequence cross-link. each side chain of the three monomers involved undergoes a chemical modification.
Cross-linkers are defined in the dialog window shown in figure Figure 3.7, “ Cross-linker definition”. This dialog window is opened by clicking crosslinkers.
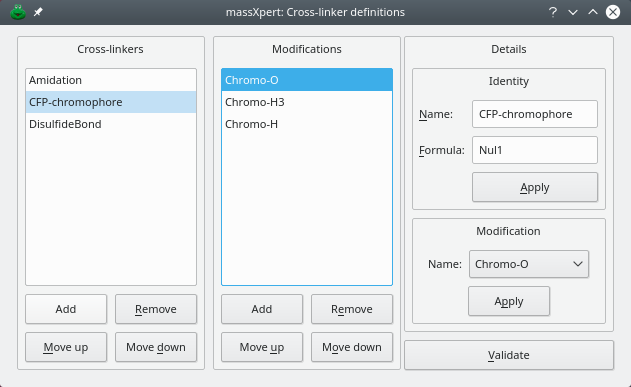
each cross-linker is defined using a name, a formula and either no modification or as many modifications as there are monomers involved in the formation of the cross-link.
Figure 3.7: Cross-linker definition #
The formation of cross-link between one or more monomers often involves chemical reactions to occur at the level of the engaged monomers. Cross-linkers defined in massxpert2 should refer to these modifications as modification objects already available in the polymer chemistry definition. Note that, in some cases, it is not necessary to define modifications to occur at the level of the cross-linked monomers.
Note
When a cross-link does not involve any specific modification, as defined in the polymer chemistry definition, then a chemical formula must be entered in the formula edit box widget, otherwise the cross-link definition will have no effect. In the figure example, the cfp-chromophore cross-linker is +nul, that is there is no chemical reaction defined for the cross-linker per se.
The example described in figure Figure 3.7, “ Cross-linker definition”, corresponds to the cross-linking reaction involved in the formation of the chromophore of the cyan fluorescent protein. That reaction involves the three following monomers: 65threonyl 66tyrosinyl 67glycyl. Each monomer undergoes a distinct chemical modification: “-o”, “-h3” and “-h”, respectively. Three modifications were thus defined: chromo-0, chromo-h3 and chromo-h, in that specific order, as these modifications are going to be sequentially applied to their corresponding monomer in the cross-linking reaction.
Warning
When multiple modifications are used, the number of these modifications must match the number of monomers involved, and their order must match the order with which the monomers are cross-linked. if no modification is defined, then, the chemical reaction that occurs upon cross-linking might be defined in the formula of the cross-linker.
3.2.4 The cleavage specifications #
It is common practice—in biopolymer chemistry, at least—to cut a polymer into pieces using molecular scissors like the following:
Proteases, for proteins;
Nucleases, for nucleic acids;
Glycosidases, for saccharides…
Note
Not only biological scissors can be defined, but also chemical ones, like cyanogen bromide, for example, that cleaves at a methionyl residue. massxpert2 allows the user to define such kind of chemical scissor.
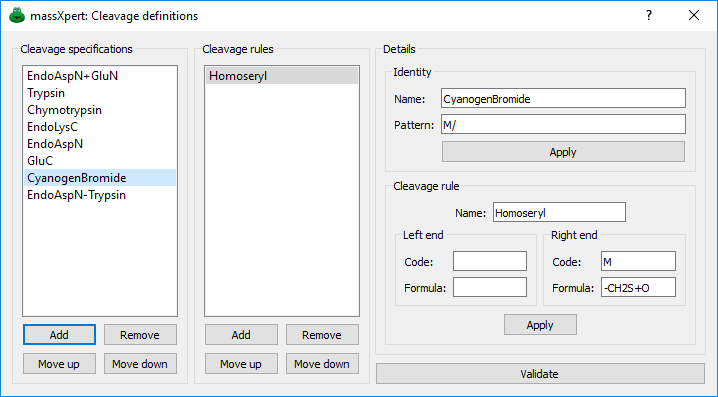
Each cleavage specification is defined using a name, a cleavage pattern and any number of cleavage rules.
Figure 3.8: Cleavage specification definition #
For each different polymer type, the molecular scissors are specific. indeed, a protease will not cleave a polysaccharide. This is why cleavage specifications belong to polymer chemistry definitions. In the example of figure Figure 3.8, “ Cleavage specification definition”, the definition of the cyanogenbromide cleavage specification is detailed (this organic reagent cleaves right of methionyl residues). The cyanogenbromide cleavage specification is qualified as so:
cyanogenbromide: the name of the cleavage agent;
m/: the sequence pattern recognized by the cleaving agent. In this case, the cleavage agent cleaves the protein right after m residues;
The cleavage rule groupbox allows the user to define the cleavage rules that might be added to the cleavage specification. The case of the cyanogen bromide reagent is interesting in this regard:
left code and left formula are two line edit widgets for the special cases of cleaving agents that not only cut a polymer sequence (usually it is a hydrolysis) but that also modify the substrate in such a way that must be taken into account by massxpert2 so that it computes correct molecular masses for the resulting oligomers. These rules are optional. However, if left code is filled with something, then it is compulsory that left formula be filled with something valid also, and conversely. The cyanogen bromide/protein reaction does not involve any chemical modification (apart from the cleavage) of the monomer left of the generated peptide, so these edit wigets are left blank.
right code and right formula m and -ch2s+o3, respectively. Same explanation as above. this cleavage rule stipulates that upon cleavage of a protein using cyanogen bromide, the methionyl residue that gets effectively cleaved must be converted to a homoseryl residue. see below for a detailed explanation.
Here are some examples of more complex cleavage patterns:
trypsin = k/;r/;-k/p: “trypsin cuts right of a ‘k’ and right of a ‘r’. But it does not cut right of a ‘k’ if this k is immediately followed by a p”;
endoaspn = /d: “endoaspn cuts left of a d”;
hypothetical = t/ys; pgt/hyt; /mnop; -k/mnop: “hypothetical cuts after ‘t’ if it is followed by ys and also cuts after ‘t’ if preceded by pg and followed by hyt. Also, hypothetical cuts prior to ‘m’ if ‘m’ is followed by nop and if ‘m’ is not preceded by ‘k’”.
Note
Please, do note that the letters in the examples above correspond to monomer codes and not to monomer names. If, for example, we were defining a “trypsin” cleavage specification pattern—in a protein polymer chemistry definition with the standard 3-character monomer codes—we would have defined it this way: “trypsin = lys/;arg/;-lys/pro”.
Now comes the time to explain in more detail what the left code and left formula (along with the right siblings) are for. For this, we shall consider that we have the following polymer sequence (1-character monomer codes): “thismwillmbecutmandthatmalso”. If that sequence had been cleaved using cyanogen bromide and if the cleavage had been total,[5] that would have generated the following oligomers: “thism willm becutm andthatm also”. but if there had been partial cleavages, one or more of the following oligomers would have been generated: “thismwillm becutmandthatm also willmbecutm andthatmalso” and so on…
Now, the biochemist knows that when a protein is cleaved with cyanogen bromide, the cleavage occurs effectively right of monomer “m” (this we knew already) and the “m” monomer that underwent the cleavage is changed from a methionyl residue to an homoseryl residue (this chemical change involves this formula: “-ch2s+o”). Amongst all the oligomers generated above, there are two oligomers that should not undergo the cleavage rule “-ch2s+o”: “also” and “andthatmalso”. indeed, these two oligomers were generated by the “cyanogenbromide” cleavage, but were not actually cleaved at the right side of a methionyl residue, because they correspond the the right end terminal part of the protein sequence (even if one them does contain a “m” residue; the cleavage did not occur at that residue).
This example should clarify why the definition clearly stipulates—in the cleavage specification for “cyanogenbromide”—that the oligomers resulting from this cleavage should “undergo the ‘-ch2s+o’ formula only if they have a ‘m’ as their right end monomer code”. These cleavage rules need to be defined in a very careful way: imagine that—in some experiments involving cyanogen bromide —that reagent would cleave right of “c” (cysteine) residues, but with no chemical modification of the “c” monomer[6]. In this case, it would be suitable to put the flexibility of massxpert2 at work by specifying that the generated oligomers should ``undergo the “-ch2s+o” formula'' only if they have a “m” as their right end monomer, so that “c”-terminated oligomers are not chemically modified. Thus the cleavage pattern might be safely defined: “m/;c/”…
3.2.5 The fragmentation specifications #
The specification of fragmentation events in a polymer chemistry definition is not a trivial task. In this section three different cases will be described, from simple to more complex. One major rule is that the fragmentation specification should be crafted in such a manner that the resulting fragment is neutral. The ionization of the fragments will be then automatically performed by massxpert2 upon calculation of the ion products according to the current ionization rule. This is a major improvement over previous versions, that forced the user to define fragmentation specifications by assuming a product ion of a given ionization ([m+h]+ for proteins or [m-h]- for nucleic acids, for example).
3.2.5.1 Simple fragmentation patterns #
One simple example of polymer chain fragmentation is the formation of a fragments with a nucleic acid (dna, in this example). Bond cleavage occurs right before the sugar-carbon-linked oxygen of the phosphoester bond linking one deoxyribonucleotide to the next. Thus, the molecular weight of the fragment corresponds to the sum of the monomer masses from the left end of the polymer up to and including the monomer being decomposed less one oxygen. note that this specification yields a nucleic acid ion product that is protonated. we thus need to remove a proton to change its charge to 0, thus the formula of the a fragmentation pattern is “-oh” this is illustrated in figure Figure 3.9, “ Fragmentation rule definition”: the name of the specification for fragmentation pattern “a” is a; the formula associated to this fragmentation pattern is -oh; the fragment encompasses the le (for “left end”) of the polymer chain; the monomer value is set to 0, which will be explained later.
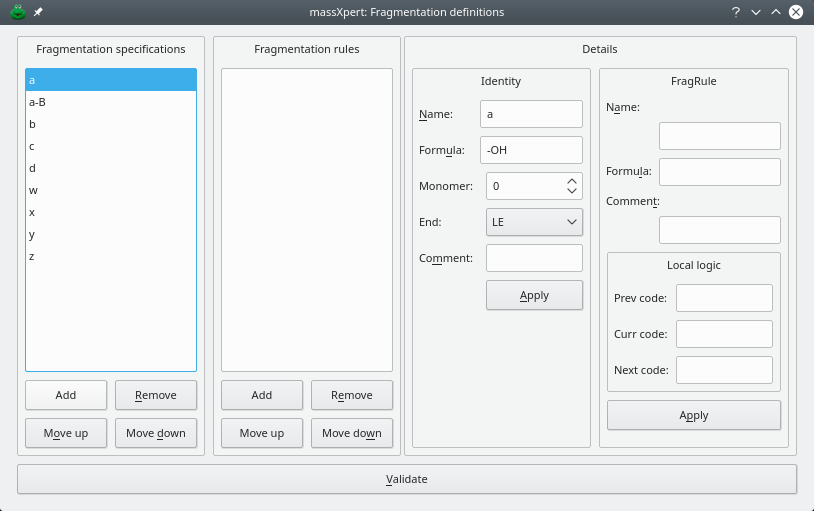
Each fragmentation rule is defined using a name, a formula and a local logic, that is a set of logical conditions which must be verified for the fragmentation specification to be applied to the fragment.
Figure 3.9: Fragmentation rule definition #
3.2.5.2 More complex fragmentation patterns #
In nucleic acids gas-phase chemistry, it often happens that not only fragmentation occurs at the level of the phospho-ribose skeleton, but also at the level of the nucleic base. These fragmentation patterns are called abasic patterns. The decomposition of the base occurs at the monomer position where the fragmentation occurs. For example, if a “atgc” oligonucleotide is fragmented according to pattern a but with nucleic base decomposition, and that fragmentation occurs at position 1, then the computation of the mass should occur like represented in figure Figure 3.10, “ Fragmentation definition with generic specification”. This figure illustrates a number of things, amongst which some known basics. the top left panel show what the configuration would be in the fragmentation definition window for this kind of fragmentation. The top right panel shows the basic constituents of the dna polymer chemistry definition: the caps are oh on the left end and h on the right end; the circled formula is the skeleton (also called backbone) and the base attached to the deoxyribose ring identifies the nucleotide. that base might be adenine, guanine, cytosine, thymine. in the “dna” polymer chemistry definition, the monomers are made of the skeleton (formula c5h8o5p) plus the formula of the base, which is understandable. The following paragraphs detail two ways to configure a base-loss fragmentation pattern.
Using a monomer-generic specification. Now, if we want to compute the mass of the a-b#1 fragment, that is fragmentation occurs according to pattern a right after the “a” monomer plus decomposition of the base (in our case this is an adenine, see figure Figure 3.10, “ Fragmentation definition with generic specification”). note that the decomposition of the base is accompanied by the formation of an insaturation on the sugar moiety of which the net formula is -h. We thus have to:
Apply an adapted specification for a fragments: removal of the h due to the insaturation on the sugar and also removal of the oh related to the formation of an a fragment: the -hoh component of the formula;
Remove one full monomer with monomer set to -1 (this equals to the removal of both the skeleton and the side chain—the adenine, here);
Add back the skeleton: the +c5h8o5p component of the formula;
Add the left end cap, since a fragments start at the left end of the fragmented polymer sequence: le (“left end”). That le bit of information will be transformed into a +oh formula, since this is the formula of the left end cap for nucleic acids.
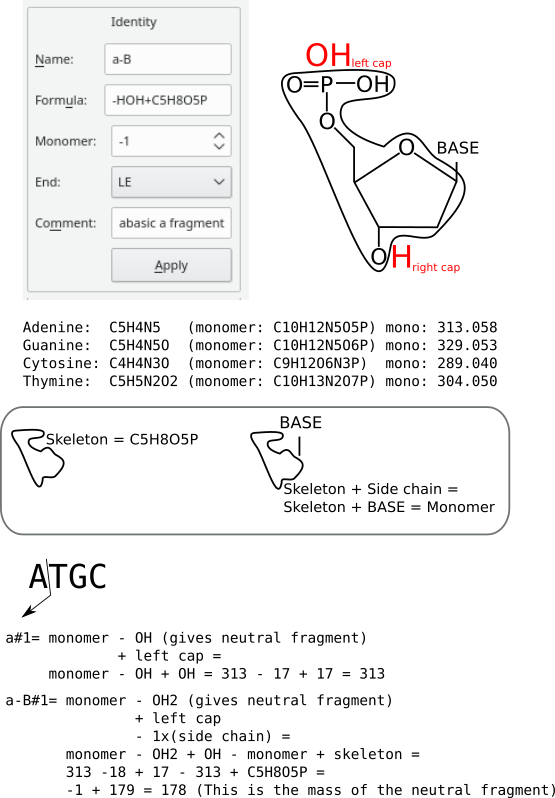
Fragmentation patterns that involve the decomposition of the nucleic base need specific configuration adjustments. Here, the removal of the nucleic base is done by first removing the whole monomer and then readding the skeleteon.
Figure 3.10: Fragmentation definition with generic specification #
The advantage of working this way is that we need not specify a fragmentation rule for each different monomer in the sequence (see below, for how this might be done). Indeed, by specifying monomer to be -1, we indicate—without knowing the monomer identity—to the mass calculation engine that once the fragmentation has occurred in the polymer chain, the mass of the monomer that got fragmented should be subtracted from the fragment mass. That subtraction removes, however too much material, as we do not want to loose the skeleton, we only want to loose the base (adenine, in our example). This is why we ask in the fragmentation specification formula that the skeleton be added (the +c5h8o5p component of the formula). Because the skeleton does not change along the polymer chain, even if the base itself changes, this computation method is generalizable, and because of this the polymer chemistry definition works.
This whole process of defining a fragmentation pattern that needs to “know” what monomer is being fragmented so as to compute the fragment masses correctly, can be performed by using fragmentation rules. This is described below.
Using a monomer-specific specification. Another way of achieving what was described above is by using fragmentation rules, whereby the fragment's mass computation is made conditional to one or more conditions that should be verified. figure Figure 3.11, “ Fragmentation definition with specific rules” shows how the a-b fragmentation pattern might be defined using fragmentation rules.
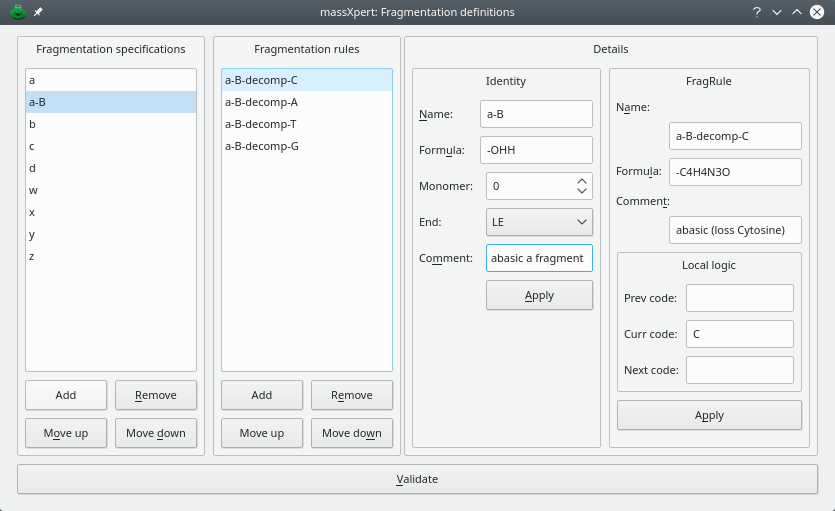
The fragmentation for pattern a with decomposition of the nucleic base at the location of the nuclotide undergoing decomposition is defined using a name, a formula and a “local logic”, that is, a set of logical conditions which must be verified for the fragmentation rule(s) to be applied to the fragment.
Figure 3.11: Fragmentation definition with specific rules #
The a-b fragmentation specification comprises 4 rules, one rule for each available monomer in the “dna” polymer chemistry definition: “a”, “t”, “g” and “c”. The figures illustrates the definition of the fragmentation specification a-b which stipulates that the mass of the fragment should be computed this way:
For the fragmentation specification part, everything is like for fragments of type a, that is, the formula is merely -ohh and the end is le (see above, for explanations);
But there is one rule (a-b-decomp-c) which adds some local logic for the fragmentation specification: the formula “-c4h4n3o” should be applied upon calculation of the fragment's masses if the monomer at which the fragmentation actually occurs (curr code) is c, i.e., if it is a cytosine. The “-c4h4n3o” formula is the formula of cytosine (the nucleic base, not the monomer).
The other rules (for curr code values a, t and g are identical to the a-b-decomp-c one unless the curr code is “a”, “t” or “g” and the formula to be removed is the formula of the corresponding dna base.
The fragmentation rule-based definition of fragmentation pattern a-b yields identical results as for the more generalizable method described earlier ( Using a monomer-generic specification).
3.2.5.3 Even more complex fragmentation patterns #
Note that in saccharide chemistry, the fragmentation patterns are extremely complex, and often totally depend on the nature of the monomers local to the fragmentation site. For example, the fragmentation behaviour at position “e” in a sequence “dear” might be different than in a sequence “dera”. massxpert2 had to be able to model these complex situations, and this is done using fragmentation rules where the local logic involves defining the prev code and/or the next code for a given curr code at which the fragmentation occurs. For example, one specific fragmentation pattern for fragmentation at “e” in sequence “dear” might be defined this way:
prev code: d;
curr code: e;
next code: a.
Instead of that fragmentation rule, one would have for fragmentation at “e” in sequence “dera” the following rule:
prev code: d;
curr code: e;
next code: r.
Note the change for next code, from a to r. Also, be aware that the “prev”, “curr” and “next” notions are polar, that is, they depend on the value of end (that is le or re). For example, if we wanted to model the fragmentation pattern at “e” for a fragment of end re, similar to what was done above with sequences “dear” and “dera”, we would have set the local logical like this:
For sequence “dear”:
prev code: a;
curr code: e;
next code: d.
For sequence “dera”:
prev code: r;
curr code: e;
next code: d.
This highly flexible fragmentation specification allows for definition of highly complex fragmentation behaviours of biopolymers.
3.3 Saving the definition #
Once the polymer chemistry definition is completed, the user can save it to an xml file. Prior to actually writing to the file, the program checks the validity of all the chemical entities in the definition. this check can be triggered manually by clicking the validate. If an error is found, it is reported so that the user may identify the problem and fix it.
The location where the file should be saved, and the manner that it may be made available to massXpert is to be described in a later chapter. It is, in fact, very important that massXpert knows where to find newly defined polymer chemistries so as to be able to use them when sequences of that polymer chemistry are created or used.
[4] Allowing more than one letter to craft monomer codes might seem trivial at first. But that design decision triggered the requirement for non-trivial algorithms throughout all the code of the of program. this is easily understandable at least in the polymer sequence editor: how are monomer codes keyed-in if “a” and “ala” are valid monomer codes in a polymer chemistry definition? the magic is described in the chapter about XpertEdit,
[5] Cleavage occurs at every possible position, right of each monomer “m”.
[6] This is a purely hypothetical situation that i never observed personally.